Formalisierte Risikobewertung von Hilfsstoffen – Ein praxisorientierter Leitfaden für Arzneimittelhersteller
Die Anforderungen an die pharmazeutische Industrie steigen stetig – nicht nur in Bezug auf Wirkstoffe, sondern auch auf sogenannte Hilfsstoffe. Diese Substanzen, die weder Wirkstoff noch Verpackungsmaterial sind, spielen eine zentrale Rolle für die Qualität und Sicherheit von Arzneimitteln. Die EU-Leitlinie 2015/C 95/02 verpflichtet Hersteller von Humanarzneimitteln seit März 2016 zur Durchführung einer formalisierten Risikobewertung aller eingesetzten Hilfsstoffe. Bereits seit 2013 ist dies in der AMWHV gesetzlich verankert. Sobald die Risikobewertungen für die Hilfsstoffe vorliegen, können diese auch für die Einführung von periodischen oder stichprobenartigen Kontrollen (Skip-Lot-Testing) der Wareneingangsprüfung der Hilfsstoffe herangezogen werden, wodurch der Prüfaufwand und damit die Kosten reduziert werden.
Warum eine Risikobewertung von Hilfsstoffen notwendig ist
Obwohl die Qualität der Hilfsstoffe in den Zulassungsunterlagen festgelegt ist und Lieferanten meist qualifiziert sind, fehlt häufig eine systematische Bewertung der Risiken, die von diesen Stoffen ausgehen können. Diese Bewertung muss nicht nur die Herkunft und Funktion des Hilfsstoffs berücksichtigen, sondern auch die Art des Arzneimittels, in dem er verwendet wird. Die Herausforderung: Viele dieser Informationen stammen aus der Entwicklungsphase und sind in Produktionsstätten oft nicht verfügbar.
Das empfohlene Vorgehen: Drei Schritte zur Risikobewertung
Die Leitlinie schlägt ein strukturiertes Verfahren vor, das auf den Prinzipien des ICH Q9 „Quality Risk Management“ basiert:
- Bestimmung der angemessenen Guten Herstellungspraxis (GMP) – abhängig von Art und Verwendung des Hilfsstoffs.
- Ermittlung des Risikoprofils des Herstellers/Lieferanten – unter Berücksichtigung von Zertifikaten und Produktionsbedingungen.
- Bestätigung der Anwendung der angemessenen GMP – durch Audits, Fragebögen oder Dokumentenprüfung.
Priorisierung als Schlüssel zur Effizienz
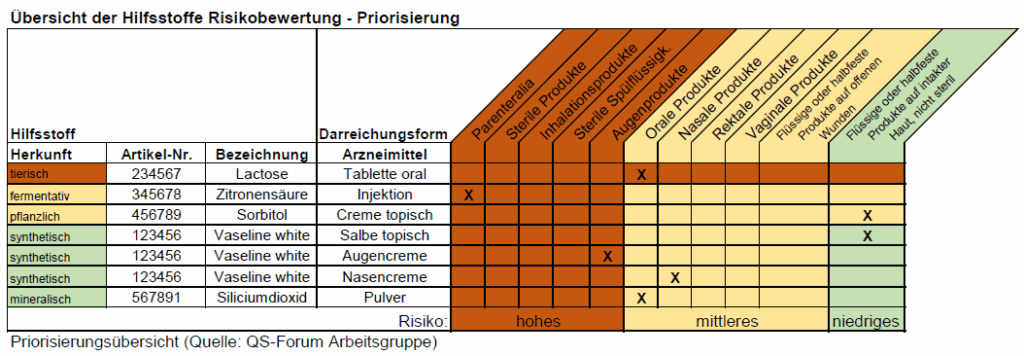
Da viele Hersteller über ein breites Portfolio mit teils über hundert Hilfsstoffen verfügen, empfiehlt sich eine Priorisierungsmatrix. Diese klassifiziert Hilfsstoffe nach Risiko für den Patienten:
- Hohes Risiko: Tierische Herkunft, Einsatz in sterilen Produkten, Parenteralia, Inhalationspräparate, Augenprodukte.
- Mittleres Risiko: Pflanzliche Herkunft, Fermentationsprodukte, Einsatz in oralen, rektalen, vaginalen oder nasalen Arzneiformen.
- Geringes Risiko: Mineralische oder synthetische Herkunft, Anwendung auf intakter Haut.
Die Bestimmung der Guten Herstellungspraxis anhand der Art und Verwendung der Hilfsstoffe kann in die folgenden zwei Schritte gegliedert werden:
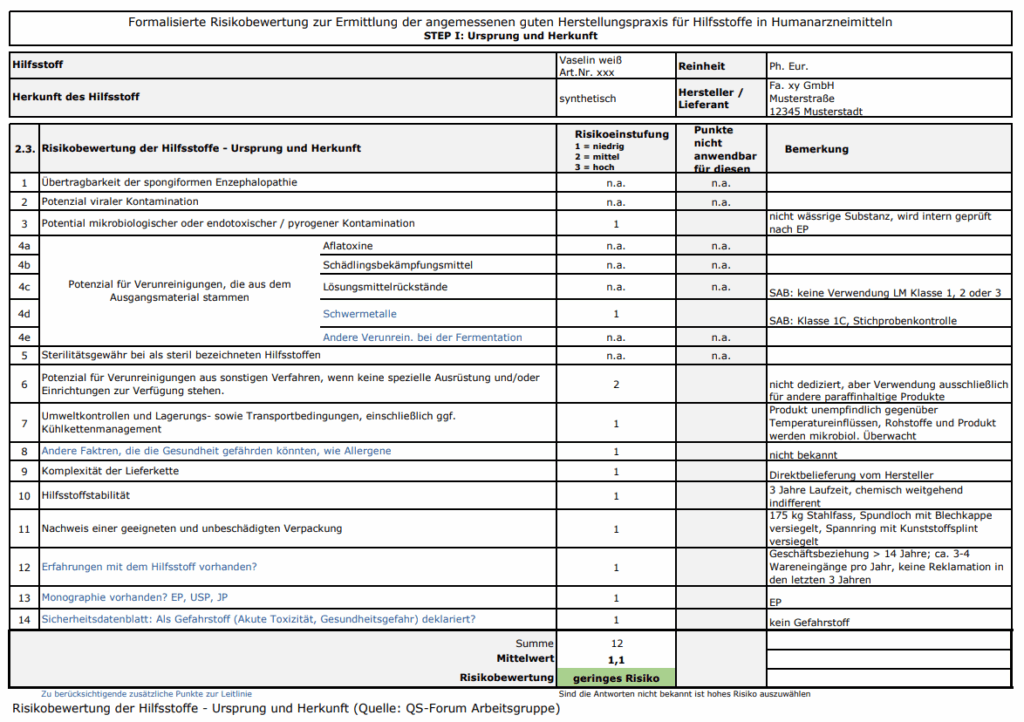
Teil I: Ursprung und Herkunft des Hilfsstoffs
Hier werden potenzielle Risiken wie BSE-Übertragbarkeit, virale und mikrobiologische Kontamination, Rückstände von Lösungsmitteln oder Pestiziden sowie die Komplexität der Lieferkette bewertet. Die Bewertung erfolgt numerisch (1 = niedrig, 2 = mittel, 3 = hoch), und das arithmetische Mittel ergibt die Gesamtrisiko-Einstufung.
Teil II: Verwendung und Funktion des Hilfsstoffs
Dieser Abschnitt berücksichtigt die Darreichungsform, die Funktion des Hilfsstoffs (z. B. Konservierungsmittel, Emulgator), die tägliche Aufnahme durch den Patienten und bekannte Qualitätsmängel. Auch hier erfolgt eine numerische Bewertung mit anschließender Gesamteinstufung.
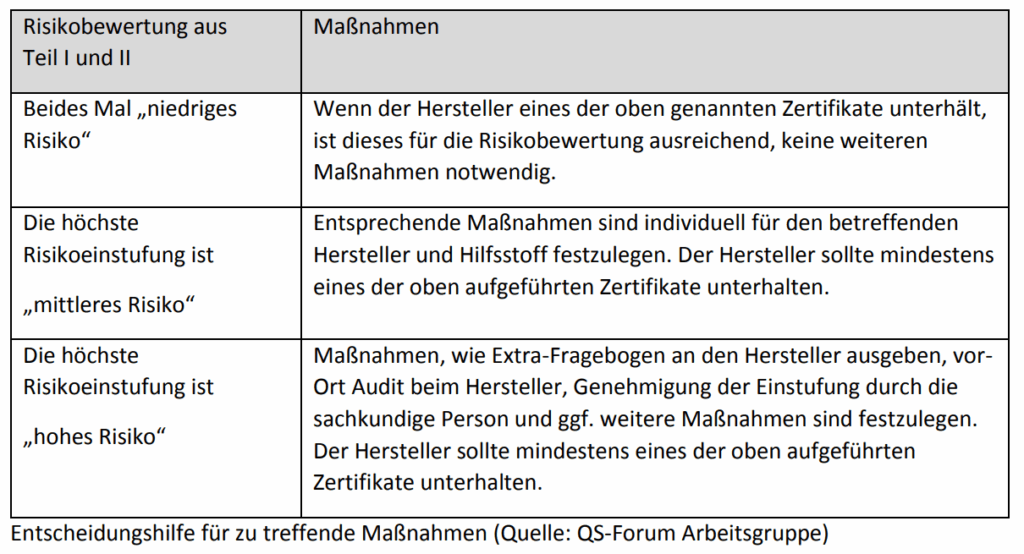
Bewertung des Herstellers/Lieferanten
Ein Hersteller mit Zertifikaten wie EXCiPACT, ISO 9001 oder einem GMP-Zertifikat gilt als vertrauenswürdig. Liegen keine Zertifikate vor oder ist die Risikoeinstufung des Hilfsstoffs hoch, muss eine Gap-Analyse durchgeführt werden. Dabei werden die Anforderungen der Guten Herstellungspraxis mit den tatsächlichen Gegebenheiten beim Hersteller verglichen.
Bestätigung der GMP-Anwendung
Die sicherste Methode ist ein Audit beim Hersteller – jedoch oft kostenintensiv und nicht immer möglich. Alternativ können Fragebögen oder telefonische Interviews genutzt werden. Die Ergebnisse fließen in eine Bewertung der GMP-Konformität ein.
Maßnahmenplan und Risikominimierung
Bei unzureichender GMP-Konformität müssen Maßnahmen definiert werden – von zusätzlicher Analytik über Sterilisation bis hin zum Austausch des Hilfsstoffs. Letzteres kann jedoch zu Zulassungsänderungen und Bioäquivalenzstudien führen.
Risikokommunikation und laufende Überprüfung
Die Bewertung muss intern und extern kommuniziert werden – etwa mit Qualitätskontrollabteilungen oder Lieferanten. Zudem ist eine regelmäßige Überprüfung notwendig, z. B. bei Änderungen im Herstellungsverfahren, neuen Risiken oder neuen Arzneimitteln. Die Integration in bestehende Systeme wie Lieferantenqualifizierung oder Product Quality Review wird empfohlen.
Fazit
Die formalisierte Risikobewertung von Hilfsstoffen ist kein bürokratisches Hindernis, sondern ein strategisches Instrument zur Sicherstellung der Arzneimittelqualität. Sie ermöglicht eine transparente, nachvollziehbare und patientenorientierte Bewertung und sollte bereits in der Entwicklungsphase eines Arzneimittels berücksichtigt werden. Sobald die Risikobewertungen vorliegen, können sie zur Einführung stichprobenartiger Wareneingangskontrollen (Skip-Lot-Testing) unterstützend genutzt werden, um Prüfaufwand und Kosten zu senken.